
Researchers engineered protein-like polymers that replicate complex enzyme functions. This work, which was verified using X-ray characterization techniques at the ALS, offers a cost-effective, scalable approach that paves the way for functional materials in biomedicine, energy, and manufacturing. Read more »![]()

A World of Vibe Coding Opportunities at the ALS
In April 2026, a panel of vibe coders hosted a tutorial at the ALS. From a general overview to specific strategies to optimize the code, participants learned all about how vibe coding can improve their workflows. Read more »
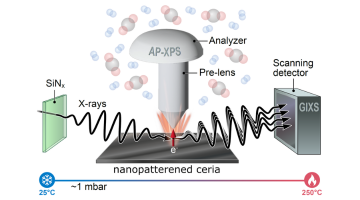
Seeing Double: Dual Measurements of Ceria Provide Insights for Catalysis
In a recent study at the ALS, researchers employed an approach that combines two techniques and uses a single X-ray beam to capture both chemical and structural changes in nanopatterned ceria during catalytic reactions. Read more »

ALS-United: Changchun Sun and Jonah Weber
ALS-United is an opportunity to meet the people collaborating at the Advanced Light Source and the ALS Upgrade Project. Hear firsthand how team science enables the cutting-edge research of today and builds the facility of the future. This month, we spoke with Changchun Sun (Staff Scientists/Engineer) and Jonah Weber (Electronics Engineer). Read more »

Poster Session for Spring 2025 Cohort of ALS Fellows
On the sunny afternoon of April 23, the ALS community gathered to celebrate a year of excellent science from the ALS fellows. The Spring 2025 Cohort presented their posters, and everyone enjoyed cake on the patio. Read more »